

系統解析とは、生物の持つDNAなどの情報を基にして、生物の進化の過程を推測する技術のことを言います。
複数の生物の進化の過程をトーナメント表のように表した「系統樹」と呼ばれる図を作成する事が多くの場合の目的になります。
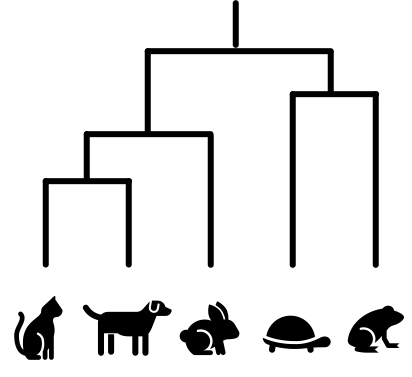
系統樹を生物の教科書などで見かけた事があるかもしれませんが、これはDNAなどの情報に基づき、数理的に計算され描かれているものがほとんどです。(地中で見つかった化石の深さなどから系統樹が作成されることもあります。)
しかし、昔の生物のDNAを採取することは不可能なので、結局は「今、存在する生物」のDNAのデータしかありません。
そこからどうやって生物進化の歴史を知る事ができるのでしょうか。
本記事ではその方法「系統解析」についての大枠をご説明します。
生物のDNA
地球上の全ての生物にはDNAやRNAといった、その生物の情報を記録する物質を細胞内に持っています。
ヒト、イヌ、ネコなどの哺乳類だけでなく、昆虫や魚、植物、ウイルス、細菌でさえも同じです。
大雑把に言えば、その細胞内の物質(以降、便宜上これをDNAと呼びます)こそが、その生物の特徴やら遺伝の情報やらを全て担っているわけです。
そしてこの物質は、決して複雑なものではなく、たった4種類の物質で構成されています。
4種類しか無い代わりに、それが何十億個も連なることによって様々なパターンを生み出しています。
DNAが生物の情報を担っているということは、近い生物のDNAの並びはやっぱり近い事になります。ヒトとチンパンジーのDNAは98%以上一致しているという報告もあります。
「ヒトとカブトムシ」より「ヒトとイヌ」の方がDNAは近いですし、「ヒトとチンパンジー」はもっと近く、「日本人とフランス人」ならおそらくもっと近いでしょう。
・・・ところで、太古「生命」というものが誕生した時、生物はせいぜい数種類しか居なかったはずです。
それが、猿人からヒト・ゴリラ・チンパンジーなどに分かれてきたように、長い歴史の中で様々な生物種に分岐してきました。
つまり、元はみんな同じDNAを持っていたのに、それが進化の過程で少しずつDNAの構成が変わってきて違う生物が生まれてくるわけです。
簡単に言えば、「現在のDNAの並びが近い生物間ほど、最近分岐した」と捉えられるわけです。
(生物進化の仕方についても複数の意見がありますが、今回はそのあたりの話は割愛します。)
先の例で言えば、「ヒトとカブトムシ」が分岐したのは遥か昔ですが、「日本人とフランス人」が分岐したのは生命の歴史からすれば最近の事と推定されるわけです。
シンプルながらも非常に大量なこのDNAの情報こそが、生物の進化の歴史を辿るための重要な”ビッグデータ”となります。
それでは、そのビッグデータを用いて具体的に進化の歴史を辿る手順を見ていきましょう。
DNAデータの収集
何はともあれ解析したい生物のDNA情報を集めなければいけません。
わざわざ自分で生物から抽出してくるなどと言うことは無く、既にWeb上で無償公開されているデータを引っ張ってきます。
有名なサイトでは「DDBJ」「GenBank」などがあります。
また、何十億にものぼる配列全てを分析に使う、ということも通常ありません。
DNAにも様々な「領域」があり、解析したい生物の「ある領域」のデータに絞って分析します。
この「領域」も、まったく進化や遺伝に関係ない部分を使っても仕方ないので、きちんと選定する必要があります。
といっても、対象の生物などによっても最適な領域は変わりますので、一概に「この領域を使いましょう!」とは言えません。
ただ、16S rRNAや23S rRNA領域を用いると正確性の高い系統解析ができると言われています。
使う領域を決めたら先述したサイトでデータを取得してきます。
データの形式も「FASTA」「PHYLIP」「NEXUS」など、幾つか種類があります。
最も広く使われているのはFASTA形式かと思われますが、ツールによってはFASTA形式を受け付けないものもあります。
どんなツールで解析を行うかによって変わりますので、きちんと使う予定のツールの受け入れ可能な形式は調べておきましょう。
系統樹作成のための前準備(アライメント)
さて、系統解析したい生物のデータが集まったら、いよいよ解析・・・ですが、前準備があります。
1つ分析するにあたって問題となるなのが、「生物によってDNAの長さはまちまち」ということです。(近い生物同士ならば長さが同じという事もあります。)
なぜ生物によって配列の長さが変わってしまうかと言えば、進化の過程でDNAが変異するだけでなく、「挿入」や「欠失」という変化も起こるからです。
系統解析においては、多くの場合、長さがばらばらだと分析が難しいため配列の長さを揃える作業が必要です。
この作業のことを「アライメント」と呼びます。
例えば、「ACTG」という配列と「ATG」という配列があったら、後者を「A-TG」とすれば長さが合う上に、2つの配列の並びが近くなります。
このように、闇雲に長さを揃えれば良いのではなく、なるべくデータ全体がバラバラにならないように空白(ギャップと呼ばれます)を入れ、長さを揃えます。
(このあたりの話は進化距離の記事に、もう少し詳しく記しています。)
この作業を手作業で行うのはほぼ無理なので、既存のアライメント用アルゴリズムを利用して自動で行います。
これにも「ClustalW」「MAFFT」「T-Coffee」など様々な種類があり、どれも一長一短があります。
しかも、それぞれのメソッド内にも様々なハイパーパラメータがあります。
使うメソッドやパラメータにより得られる結果は異なりますが、進化の過程が分からない以上、どのアライメント結果が正解なのかも分かりません。
結果を人間が見て違和感が無い結果なら一先ずはOKと考えます。
(データ量が少なければ、人間が結果を見て微調整する場合もあります。)
系統樹の作成
さて、これでいよいよ系統樹が作成できます。
DNAの配列を見て、どの生物同士がどれほど近いかを調べ、その情報を基に全データの関係性を連結して一つの「系統樹」を作っていきます。
機械学習の用語を使えば、まさに「階層型クラスタリング」そのものです。

しかし、今回は対象が生物のDNAデータということで、従来機械学習で良く使われるクラスタリング手法をそのまま適用することは稀です。
系統解析のために生まれた階層型クラスタリング手法が幾つかあり、大別すると以下に分けられます。
距離行列法
様々な場面でで良く使われるクラスタリングに近い方法です。
データから全てのペア間の距離を計算し、その距離が小さい方からデータをくっつけていきます。
生物が100種類あったら、\(_{100}C_{2}\)で4950個の距離を計算する必要があります。
また、この距離の計算にはDNA配列専用の計算が必要です。
確かに、「ACGTTCGTTC」と「CATTACGTAC」の距離は?と言われても難しいですね。
この、DNA配列専用の距離を「進化距離」や「遺伝的差異」と呼びます。
進化距離にも多数あるので、これも何かひとつ選ばなければいけません。
詳しくは進化距離の記事をご覧ください。
また、クラスタリング手法も幾つかあります。
一般的な階層型クラスタリングでも用いられるUPGMA法などを適用する場合もありますが、系統解析においては「近隣結合法(NJ法)」を使うのがメジャーです(2019年現在)。
通常のクラスタリングはどの要素が近いか遠いかのみ分かればOKな事が多いのですが、系統解析ではそれだけでなく「枝の長さ(=分岐してからどれほどの年月が経ったか)」が重要になってきます。
NJ法は、データを極力、枝の長さに反映させたクラスタリング手法となっています。
距離行列法を用いて系統解析できるツールには、「MEGA」「Phylip」などがあります。
最大節約法
最大節約法は、距離行列法のようにペア間の距離を逐一計算するのではなく、配列全体のデータを一気に見ます。
系統樹を少しずつ構築してゆくのではなく、まず考えられる樹形を全パターン作ってしまいます。
その後で、それぞれの樹形がどれほど信憑性のあるものか?・・・という計算を行い、最も信憑性の高い樹形を探します。
生物種数が多くなると、いくらコンピュータに計算させると言っても総当りで樹形を見ていくのは大変です。
なので、全部の樹形を調べるのではなく、正解にアタリを付けてその周辺の樹形を集中的に見ていく「枝刈り」の手法を取ることもあります。
(距離行列法で系統樹を書き、その系統樹を少しいじって作れる樹形だけを調査対象にする、など)
\(n\)生物種における考えられる樹形は
\[\prod_{i=1}^{n-2} (2i+1)\]
で計算できます。3生物なら考えられる樹形は3通り、4生物なら15通り、5生物なら105通りになります。
最大節約法の考え方はシンプルで、ある樹形について、最低、何回の置換があればこの系統樹が発生するか?を計算します。
全ての樹形についてその計算を行い、最も「置換回数」が少なくて済んだ系統樹が、最節約法における「正解」となります。
このように、置換の回数を見るだけなので、最節約法では分岐の順番は分かりますが、系統樹の枝の長さは分からない事にご注意ください。
最大節約法を用いて系統解析できるツールには、「PAUP*」「Phylip」などがあります。
最尤法
実際の進化は、一定のペースで進むわけではなく、生物種によってまちまちです。
最尤法は確率論の考え方を取り入れることによって、生物により進化のスピードが違う問題を解消した系統樹作成方法です。
よって、完成する系統樹は枝の長さまで含めて計算されます。
計算はやや複雑となるため、最節約法より更に時間が掛かります。よって、最節約法の章で述べた枝刈りの手法と併用される事がほとんどです。
最尤法を用いて系統解析できるツールには「PAUP*」「RAxML」などがあります。
ベイズ法
ベイズ法も最尤法と同様に、確率論を用いて系統樹を作成する手法ですが、「ベイズ確率」という確率を用いて系統樹作成を行います。
最尤法とベイズ法のどちらが優れるかについては流派が存在しており、一概に優劣は言えません。
ベイズを用いて系統解析できるツールには「MrBayes」「PAML」などがあります。
進化の推測
さて、系統樹が書けたらいよいよ生物進化の歴史が浮き彫りになってきます。
例えば、仮に以下のような系統樹が作成されたとしましょう。

枝の長さがうまく「分岐してから経過した時間」を表現しているとすれば、この枝の長さの比率が、そのまま年代の長さと比例していると仮定できます。
(つまり、最大節約法では分岐の順番は分かりますが枝の長さは分かりませんので、分岐年代の推測には使えない事になります。)
「ヒトとイヌ」がいつ分岐したのかを正確に考えるのは難しそうです。
しかし「ヒトとチンパンジー」ならば、化石が見つかった層の深さなどの情報から、ある程度「事実ベース」で年代が推測できます。
例えば、ヒトとチンパンジーが分岐した年代を500万年前としましょう。
系統樹における「ヒトとイヌ」の枝の長さが「ヒトとチンパンジー」の12倍だったとしたら、「ヒトとイヌ」が分岐したのは6,000万年前ころではないかと推定することができます。
そういう意味で、クラスタリングの「枝の長さ」というものが系統解析ではとりわけ重要になってくるわけです。
(研究テーマによっては分岐の順番さえ分かれば良い、という事もあります。)
系統解析の応用
もともとは生物の分岐年代を推測するために生まれた手法ですが、別の分野に応用する事例も見られています。
例えば、単語の並びを見て言葉の進化を調べる言語の研究。
いつの時代に、感謝の言葉が「ありがとう」と「おおきに」に分かれたのか?などを、文化的な側面だけでなく、理論的な側面からも調査することができます。
おわりに
以上が、系統解析についての概略でした。
系統解析については、以下の書籍などにも丁寧に解説されています。
本記事に記した内容が数学的に丁寧に解説されており、現在販売している日本語の書籍の中では、最も充実しているのではないかと考えています。

理論面はそこそこにして、概要をざっと掴みたいのであれば、以下の書籍も読みやすく、おすすめです。

